Tailored Gene Therapy: A Landmark for Rare Baby Disorder
A groundbreaking medical achievement has emerged, offering unprecedented hope for infants born with rare, life-threatening genetic conditions.

Tailored Gene Therapy: A Landmark for Rare Baby Disorder
For the first time, physicians have successfully administered a highly customized gene-editing therapy to a baby suffering from a unique genetic mutation. This bespoke treatment, specifically engineered to repair the child’s distinct defect, represents a significant leap forward in personalized medicine and holds profound implications for the future of rare disease treatment.
The Patient’s Journey: Diagnosing a Life-Threatening Disorder
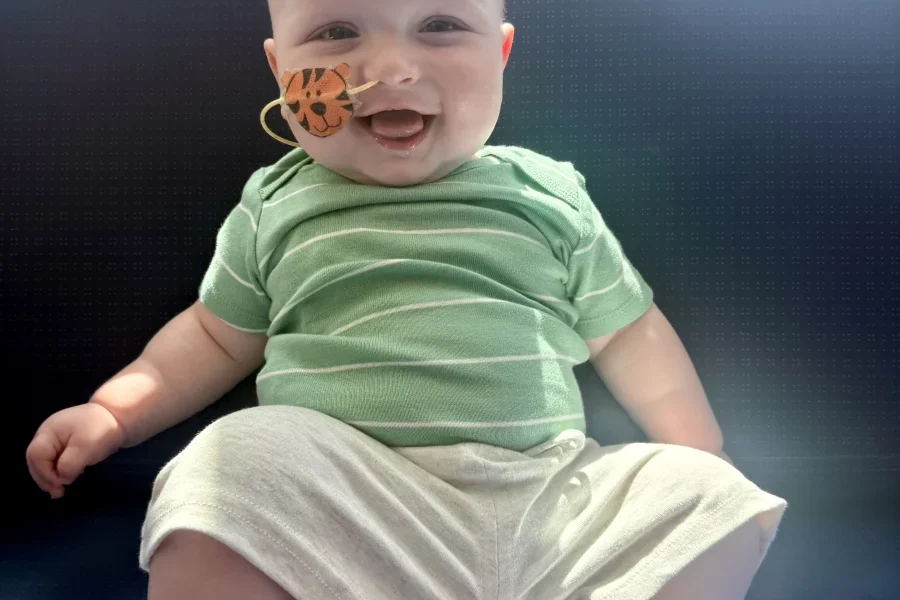
The patient at the center of this medical marvel is KJ Muldoon, who was born at the Children’s Hospital of Philadelphia (CHOP) in August (presumably 2024, given the article’s context). Initially appearing healthy, KJ’s doctors soon recognized urgent signs of a serious underlying condition.
As his father, Kyle Muldoon, recounts, the medical team quickly informed the parents of their son’s critical state, setting the stage for a race against time to identify and treat his debilitating illness.
Understanding Carbamoyl Phosphate Synthetase 1 (CPS1)
KJ was diagnosed with Carbamoyl Phosphate Synthetase 1 (CPS1) deficiency, an inherited genetic disorder belonging to a group known as urea cycle disorders. This condition prevents the body from properly processing protein, leading to a perilous accumulation of toxic ammonia in the child’s bloodstream.
Elevated ammonia levels pose a severe threat, making affected infants highly susceptible to irreversible brain damage and potentially even death. While medications can partially manage ammonia levels, and a liver transplant offers a more definitive solution, the latter is typically not an option until a child reaches at least one year of age. Tragically, by this time, many patients have already suffered irreversible neurological harm, underscoring the urgent need for early, effective intervention.
The Innovative Base-Editing Treatment Approach
Faced with KJ’s dire prognosis, a dedicated team of researchers and clinicians embarked on a pioneering therapeutic path. Led by experts like Dr. Rebecca Ahrens-Nicklas, a pediatrics and genetics assistant professor at CHOP and the University of Pennsylvania, and Dr. Kiran Musunuru, a translational research professor at the University of Pennsylvania, the team developed a gene-editing therapy using a cutting-edge technique known as “base-editing.”
This sophisticated method allowed for precise, molecular “surgery” on KJ’s genetic code, literally rewriting the faulty sequence in his liver cells to correct the defect. The treatment involved three infusions, delivering billions of microscopic gene-editors directly to the target mutation.
Overcoming Challenges: From Research to Real-World Application
The path to treating KJ was fraught with scientific and logistical hurdles. The research team had previously undertaken similar “dress rehearsals” attempting to develop gene-editing therapies for six other children with rare disorders, though these earlier efforts did not yield timely solutions. However, this prior experience honed their skills, allowing them to formulate a specific gene-editing solution for KJ within weeks of his birth.
Given the immediate danger to KJ’s life, the Food and Drug Administration (FDA) granted an emergency exception from standard testing protocols, allowing the experimental treatment to proceed. This presented KJ’s parents with an “impossible decision”—opt for an unproven therapy or face the grim alternatives. Despite the immense pressure, the Muldoons courageously chose the novel treatment, administered on February 25.
The medical team described the day as both “nerve-wracking” and “momentous,” filled with apprehension as they delivered a drug never before given to an infant in such a personalized context.
Ethical Considerations in Pioneering Pediatric Treatments
The application of a highly experimental treatment on a child, particularly one facing a life-threatening condition, naturally raises significant ethical considerations. Independent bioethicists, including Dr. Lainie Ross from the University of Rochester and Laurie Zoloth from the University of Chicago, reviewed KJ’s case for NPR.
They affirmed that the researchers appeared to have taken proper precautions, highlighting the importance of thorough discussions with KJ’s parents regarding all available options, including the possibility of a liver transplant. Dr. Peter Marks, a former high-ranking FDA official, further acknowledged the “transformational” potential of this approach in an accompanying editorial, noting it could be applicable to “hundreds to thousands of diseases.”
Initial Results: Glimmers of Hope for KJ’s Development
While caution remains paramount, the early signs following KJ’s treatment are profoundly encouraging. After an initial low-dose infusion, two subsequent infusions appear to be working effectively without any observed side effects. Doctors have successfully reduced his ammonia-reducing medication by half, and KJ can now tolerate more protein in his diet, leading to healthy weight gain.
More importantly, his parents, Nicole and Kyle Muldoon, are witnessing their son achieve developmental milestones that once seemed unattainable. Seeing him sit upright independently, wave, and roll over on his own fills them with immeasurable joy, offering concrete evidence of the treatment’s positive impact. Dr. Ahrens-Nicklas maintains a cautiously optimistic stance, stating that while “the signs are promising,” they are “still in early days.”
Broader Implications: A New Era for Rare Disease Treatment
The success of KJ’s tailored therapy is seen as a watershed moment for rare disease treatment. Dr. Kiran Musunuru believes this case signifies an “important first step towards an entirely new type of personalized medicine,” suggesting it is “the future of modern medicine.” Similarly,
Fyodor Urnov, scientific director at the Innovative Genomics Institute at UC Berkeley, hailed it as the year “CRISPR-on-demand is truly born,” envisioning a future where genetic cures can be developed much more rapidly and, crucially, at a significantly lower cost. This approach could circumvent the current economic disincentives for pharmaceutical companies to invest in treatments for ultra-rare disorders, potentially unlocking therapeutic options for millions of children globally.
Dr. Edward Neilan of the National Organization for Rare Disorders lauded it as “very significant” for patients with no existing off-the-shelf treatments.
The Path Forward: Research and Future Accessibility
Despite the immense promise, experts emphasize that much more research is needed. Andrea Gropman of St. Jude Children’s Research Hospital and Alexis Komor of the University of California, San Diego, highlight the critical need for “longer-term follow-up” to ascertain the lasting efficacy of the treatment and address other open questions.
While the cost of KJ’s specific treatment, supported by the National Institutes of Health, could not be estimated due to its research nature, the hope is that this pioneering method will establish a precedent, paving the way for more rapid and, eventually, more affordable gene-editing therapies.
The focus now turns to diligent monitoring and continued research to fully understand and expand the potential of this truly transformative medical breakthrough.

The United States is currently grappling with a alarming public health challenge: a significant resurgence of measles cases. According Read more

For individuals managing diabetes, understanding the glycemic impact of foods is paramount. The glycemic index (GI) is a numerical scale Read more

The liver is one of the most hardworking organs in the human body. It acts as the body's primary detoxification Read more

Billy Evans, partner of Theranos founder Elizabeth Holmes, has successfully raised millions of dollars to launch a new artificial intelligence Read more
A significant and concerning trend is emerging in Oregon's public health landscape: a record number of families are opting out Read more
The United Kingdom government is reportedly advancing plans to significantly expand access to effective weight loss jabs, such as Ozempic Read more